Инструкция по применению лекарств, аналоги, отзывы Фактор Виллебранда
Содержание:
Техника выполнения
Построение калибровочной кривой. Далее необходимо построить калибровочную кривую, используя линейную систему координат. Для построения калибровочной кривой используют значения оптической плотности, полученные при разведении образца с известной концентрацией фактора Виллебранда. На ось х наносят значения концентрации vWF:Ag в Ед/мл или процентах (1 Ед/мл = 100 %), а на оси у указывают поглощение. В тех случаях, когда имеются образцы, значение абсорбции которых выше, чем значение самой высокой точки калибровки, они должны быть дополнительно разведены буфером для инкубации (1+1) и повторно исследованы. При этом полученная в результате измерения концентрация должна быть умножена на коэффициент разведения.
Лечение Болезни Виллебранда
Наиболее важным патогенетическим моментом в терапии, способствующим нормализации (зачастую только временной) всех нарушенных гемостатических функций, является трансфузионная терапия – введение гемопрепаратов, содержащих комплекс фактора VIII, в том числе фактор Виллебранда.
С этой целью чаще всего используют антигемоф ильную плазму и криопреципитат.
Кроме того, под влиянием введенного извне фактора Виллебранда повышается собственная продукция фактора VIII, поэтому заместительная терапия болезни Виллебранда требует значительно более редких трансфузий и меньших доз гемопрепаратов, чем лечение гемофилии А. Однократные переливания антигемофильной плазмы или криопреципитата повышают к концу первых суток уровень фактора VIII почти до 100%, после чего его концентрация выше 50% поддерживается самостоятельно в течение 36 ч. Правда, концентрация самого фактора Виллебранда снижается раньше, в силу чего тромбоцитарно-сосудистый гемостаз вновь нарушается при еще высоком уровне VIIIk в плазме. Этим объясняется то, что при болезни Виллебранда трансфузионная терапия более стойко и надежно поддерживает уровень фактора VIII и предупреждает послеоперационные кровотечения, чем купирует микроциркуляторные кровотечения (маточные и носовые). В связи с этим наиболее целесообразно введение гемопрепаратов (антигемофильной плазмы, криопреципитата) не реже 1 раза в 2 дня и в разовой дозе не меньше 15 ЕД/кг.
Коррекция развивается постепенно, поэтому перед хирургическими вмешательствами трансфузии начинают за 2-4 дня до операции, а при родах – в самом начале родовой деятельности.
Показанием к заместительной терапии служат обильные и длительные кровотечения любой локализации, хотя при маточных кровотечениях такое лечение не всегда эффективно.
Переливания тромбоцитной массы при болезни Виллебранда неэффективны, поскольку дисфункция тромбоцитов при этой болезни вторична. По этой же причине при болезни Виллебранда оказались малоэффективными и многие фармакологические препараты, применяющиеся при тромбоцитопатиях, — АТФ, соли магния, серотонин и др.
Принципиально новым является использование в лечении болезни Виллебранда аргинин-терминального синтетического аналога вазопрессина.
При легких и среднетяжелых заболеваниях доказана эффективность аминокапроновой кислоты в дозах до 0,2 г/(кг/сут), которая применяется при всех кровотечениях микроциркуляторного типа, в том числе при маточных кровотечениях с первого дня менструального цикла до окончания менструации.
Следует избегать совместного применения аминокапроновой кислоты, противозачаточных препаратов и криопреципитата, так как такое лечение может осложниться диссеминированным внутрисосудистым свертыванием крови или тромбозами. Носовые кровотечения купируют так же, как при гемофилии.
https://youtube.com/watch?v=lp7Zvofh3QY
Диагностика и лечение болезни Виллебранда
В распознавании болезни Виллебранда важную роль играет семейный анамнез, клиническая картина и данные лабораторного скрининга сосудисто-тромбоцитарного и плазменного гемостаза. Назначается общий и биохимический анализ крови, коагулограмма с определением уровня тромбоцитов и фибриногена, времени свертывания; ПТИ и АЧТВ, проводятся проба щипка и проба жгута. Из общих обследований рекомендовано определение группы крови, исследование общего анализа мочи, анализа кала на скрытую кровь, УЗИ брюшной полости.
Для подтверждения факта болезни Виллебранда определяют уровень VWF в сыворотке крови и его активность, ристоцетин-кофакторную активность с использованием методов иммуноэлектрофореза и ИФА. При болезни Виллебранда II типа, при нормальном уровне VWF и VIII факторов, информативно исследование фактора активации тромбоцитов (PAF), активности VIII фактора свертывания, агрегации тромбоцитов. Для пациентов с болезнью Виллебранда характерно сочетание сниженного уровня и активности VWF в сыворотке крови, удлинения времени кровотечения и АЧТВ, нарушения адгезивной и агрегационной функции тромбоцитов.
Болезнь Виллебранда требует проведения дифференциальной диагностики с гемофилией, наследственными тромбоцитопатиями. Кроме консультации гематолога и генетика, дополнительно проводятся осмотры отоларинголога, стоматолога, гинеколога, гастроэнтеролога.
Регулярного лечения болезни Виллебранда с малосимптомным и умеренно выраженным гемосиндромом не проводится, но у пациентов остается повышенный риск кровотечений. Лечение назначается в случае их возникновения во время родов, при травмах, меноррагиях, гемартрозе, профилактически — до хирургического и стоматологического вмешательства. Цель подобной терапии — обеспечить минимально необходимый уровень дефицитных факторов свертывания крови.
В качестве заместительной терапии показана трансфузия антигемофильной плазмы и криопреципитата (с высоким содержанием VWF) в дозах меньших, чем при гемофилии. При болезни Виллебранда I типа для прекращения кровотечения эффективно назначение десмопрессина. При легких и среднетяжелых формах геморрагий может применяться аминокапроновая кислота, транексамовая кислота. Для остановки кровотечения из раны используется гемостатическая губка, фибриновый клей. При повторяющихся маточных кровотечениях применяются КОК, в отсутствие положительного результата выполняется гистерэктомия — хирургическое удаление матки.
Причины и факторы риска
Заболевание наследуется по аутосомно-рецессивному или аутосомно-доминантному типу. Вероятность передачи заболевания от матери к ребенку составляет 50%, но только в 1/3 случаев присутствуют клинически значимые проявления болезни.
Основная причина возникновения патологии – нарушение гена, кодирующего фактор фон Виллебранда, который находится на коротком плече 12-й хромосомы.
В результате мутации происходит ряд изменений:
- отсутствие крупных мультимеров фактора Виллебранда;
- повышение аффинности (прочности связи, сродства) фактора к тромбоцитарному рецептору GB-Ib в сочетании с отсутствием крупных мультимеров;
- снижение аффинности фактора Виллебранда к тромбоцитарному рецептору GP-Ib без нарушения мультимерной структуры;
- снижение аффинности фактора Виллебранда к фактору VIII и т. п.
Исходя из вариантов функциональных дефектов выделяются соответствующие типы болезни.
Симптомы Болезни Виллебранда
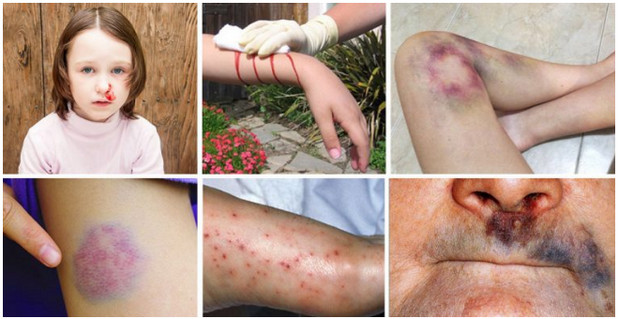
Выраженность геморрагического синдрома при болезни Виллебранда варьирует от весьма легких форм с редко наблюдающимися носовыми кровотечениями и небольшими кровоизлияниями в кожу до крайне тяжелых вариантов с очень частыми, длительными и обильными кровотечениями самой разнообразной локализации, формированием гематом и больших кровоизлияний в мягких тканях и во внутренних органах. Иногда возникают кровоизлияния в суставы.
Геморрагический синдром при I типе намного тяжелее, чем при IIА и IIB типах болезни.
Следует отметить, что интенсивность кровотечений самой различной локализации (желудочно-кишечных, маточных, носовых) зачастую не соответствует нарушению коагуляциоиного и сосудисто-тромбоцитарного гемостазов.
В частности, на фоне умеренных нарушений в этих звеньях гемостаза упорно повторяются катастрофические кровотечения какой-либо одной локализации. В подобных случаях следует думать о каких-то дополнительных местных сосудистых или стромальных дисплазиях, провоцирующих кровотечения. Для их выявления необходимо тщательное дополнительное исследование слизистых оболочек носа, зева и глотки, ротовой полости, желудка и кишечника (риноскопия, ларингоскопия, фибродуоденогастроскопия, колоноскопия). На слизистых оболочках нередко обнаруживаются сосудистые образования в виде поверхностно расположенных расширенных и извитых сосудов диаметром 1-2 мм, легко дающих обильные, трудно останавливаемые кровотечения.
Известно, что такие сосудистые образования, чаще — артериовенозные соустья, служат причиной повторяющихся желудочно-кишечных кровотечений. Данные соустья наиболее опасны при болезни Виллебранда, когда нарушены основные механизмы купирования кровотечений.
Следует отметить, что сочетание болезни Виллебранда с ангиодисплазиями и другими дефектами соединительной ткани нельзя считать случайным. У больных с этим заболеванием часто выявляется пролабирование створок митрального и других клапанов сердца, ошибочно диагностируемое без эхокардиографии как ревматический митральный порок сердца.
В таком же свете следует рассматривать сочетания болезни Виллебранда с гиперэластозом кожи, слабостью связочного аппарата (частые и привычные вывихи, разболтанность суставов, реже — синдром Марфана).
Интенсивность кровотечения имеет прямую зависимость от уровня фактора VIII в плазме, что следует учитывать как при травмах, так и при хирургических вмешательствах.
При постановке диагноза заболевания Виллебранда основываются на совокупности следующих признаков: аутосомно-доминантном наследовании заболевания, кровоточивости, значительном удлинении времени кровотечения.
К основным диагностическим признакам добавляется ряд важных функциональных характеристик, облегчающих распознавание редуцированных вариантов болезни Виллебранда.
При болезни Виллебранда выявляется постепенное, а не немедленное нарастание активности фактора VIII в плазме больных после трансфузии антигемофильной плазмы.
Коррекционный эффект трансфузии намного превосходит количество вводимого фактора VIII.
Примечательна значительно большая, чем при гемофилии, продолжительность эффекта однократного переливания — около 36 ч, что характеризуется большей продолжительностью жизни в циркуляции реципиента фактора VIII.
Наиболее информативно количественное определение фактора Виллебранда в плазме больного.
Перечисленные диагностические признаки позволяют в большинстве случаев четко аргументировать диагноз болезни Виллебранда, определять ее тяжесть и форму.
Капилляроскопические изменения (неравномерность и закрученность петель капилляров, их булавовидные расширения) в диагностике болезни Виллебранда не используют, поскольку они выявляются менее чем у половины больных и далеко не патогномоничны, однако совместно с этим они весьма демонстративны и способствуют правильной диагностике.
Аутосомно-рецессивная форма болезни Виллебранда является самостоятельным заболеванием. У гетерозигот заболевание протекает совсем или почти бессимптомно, тогда как у гомозигот наблюдается крайне тяжелая кровоточивость при почти полном отсутствии фактора VIII в плазме. Однако поражение суставов и других частей опорно-двигательного аппарата, несмотря на такой значительный дефицит фактора VIII, все же гораздо легче, чем при гемофилии.
Клинические симптомы
 Симптоматика болезни Виллебранда обуславливается снижением свертывающего потенциала крови. Основу клинического картины составляют различные по степени и виду кровотечения, причем у членов одной семьи проявления заболевания могут быть почти незаметными, а у других – летальными.
Симптоматика болезни Виллебранда обуславливается снижением свертывающего потенциала крови. Основу клинического картины составляют различные по степени и виду кровотечения, причем у членов одной семьи проявления заболевания могут быть почти незаметными, а у других – летальными.
Иногда клиника развивается в период новорожденности, а потом затухает на долгие годы, нередко заболевание диагностируется только во время операций.
Внезапность кровотечений и их нерегулярность является одной из характерных особенностей болезни Виллебранда. Наиболее часто встречаются следующие симптомы:
- кровотечения при бытовых порезах;
- носовые кровотечения;
- локализующиеся в желудочно-кишечном тракте;
- меноррагии;
- послеоперационные и послеродовые кровотечения;
- гематомы на коже;
- накопление крови в полости суставов.
Справочно. Кровотечения из слизистых рта и носовой полости нередки у пациентов детского возраста, желудочно-кишечные кровотечения могут спровоцировать прием противовоспалительных лекарственных средств и аспирина.
Тяжело протекают у таких больных язвенная болезнь и даже обычный геморрой. Повышен риск возникновения кровоточивости при травмах и инфекционных заболеваниях, потому что они повышают сосудистую проницаемость.
Предрасположенность к развитию симптомов одинакова у обоих полов, однако чаще клиника наблюдается у женщин, так как они переживают ежемесячную физиологическую кровопотерю – менструации. У заболевших женщин менструация протекает гораздо интенсивнее, её продолжительность может достигать до 10-12 дней, часто у пациенток наблюдается постоянная постгеморрагическая анемия.
Женщины с болезнью Виллебранда составляют группу повышенного риска кровотечений во время родов и в послеродовом периоде, они требуют применения различных кровосохраняющих мероприятий.
Важно! Болезнь Виллебранда может проявляться кровоизлиянием в полость сустава (гемартроз). Зачастую такая симптоматика сопровождает 2 тип заболевания.. Пораженный сустав увеличивается в размере, выглядит воспаленным, гиперемирован кожный покров над ним
Характерна резкая болезненность, чувство распирания, которые регрессируют в хронической стадии, когда суставная капсула полностью разрушена. Усугубляется течение травмированием конечности, ущемлениями близлежащих тканей, дополнительной нагрузкой на сустав при лишнем весе пациента
Пораженный сустав увеличивается в размере, выглядит воспаленным, гиперемирован кожный покров над ним. Характерна резкая болезненность, чувство распирания, которые регрессируют в хронической стадии, когда суставная капсула полностью разрушена. Усугубляется течение травмированием конечности, ущемлениями близлежащих тканей, дополнительной нагрузкой на сустав при лишнем весе пациента.
По степени тяжести клинические симптомы болезни Виллебранда можно классифицировать на три группы:
- Легкая степень (обильные менструации, петехии, долгое кровотечение из ранки);
- Средняя степень (геморрагии после чистки зубов, сдавление гематомами сосудисто-нервных пучков, появление небольшого количества крови в моче);
- Тяжелая степень (геморрагический инсульт, бронхообструкция и аспирация кровью, мезенхимальная дисплазия).
Что такое Болезнь Виллебранда —
. Впервые данное заболевание было изучено в 1926 г. ученым Виллебрандом, который на Аландских островах описал семью со своеобразным аутосомно-доминантно наследуемым геморрагическим диатезом, сходным как с тромбоцитовазопатией, так и с гемофилией. Было отмечено, что члены данной семьи страдают классической формой болезни, принадлежащей к I типу. Подтверждено доминантное наследование с разной проявляемостью патологического гена. Этим болезнь Виллебранда отличается от гемофилии, которая жестко наследуется и при которой у всех болеющих членов семьи в разных поколениях отмечается одна и та же выраженность дефицита фактора VIII.
По распространенности среди всех наследственных геморрагических диатезов болезнь Виллебранда занимает 3-е место после тромбоцитопатий и гемофилии А.
В основе патогенетических механизмов болезни Виллебранда лежит нарушение синтеза основного крупномолекулярного компонента фактора VIII, называемого также фактором Виллебранда.
По вышеперечисленным характеристикам различают «классический» тип болезни Виллебранда (тип I), при котором имеется более или менее выраженный парез синтеза рассматриваемого фактора с конкордантным снижением в плазме всех его компонентов и соответствующим отсутствием или уменьшением содержания в сосудистом эндотелии.
От этого типа отличаются вариантные формы болезни (тип II), при которых имеются качественные аномалии мультимерной структуры и свойств компонентов фактора VIII.
Болезнь по доминированию дефицита компонентов фактора VIII причисляется к плазменным дефектам гемостаза. При более углубленном рассмотрении с неменьшим правом ее можно отнести к первично-сосудистым заболеваниям, поскольку в основе болезни лежит ослабление или извращение синтеза фактора Виллебранда в эндотелии кровеносных сосудов — единственном месте его образования в организме.
Старое название болезни «ангиогемофилия» весьма близко к правильному пониманию ее сути, хотя в настоящее время редко употребляется.
Что такое болезнь Фон Виллебранда
Болезнь Фон Виллебранда — достаточно распространенное и чаще всего легкое заболевание, сопровождающееся повышенной кровоточивостью вследствие снижения уровня ФФВ без изменения его структуры или выработкой структурно измененного ФФВ с низкой активностью. Синтез ФФВ осуществляется эндотелиальными клетками и мегакариоцитами. Вначале он представляет собой полипептид из 2813 аминокислот, из которого после ряда внутри- и внеклеточных превращений, включая первичную димеризацию и полимеризацию, образуется крупный белок с молекулярной массой до 20 000 кДа. Чем выше молекулярная масса олигомеров ФФВ, тем эффективнее они обеспечивают адгезию и агрегацию тромбоцитов.
Часть ФФВ, синтезированного эндотелиальными клетками, высвобождается в кровь, а оставшаяся часть хранится в гранулах Вейбеля—Паладе, из которых он высвобождается под действием разных стимулов, например интенсивной физической нагрузки, десмопрессина или адренергических средств. ФФВ, синтезируемый в мегакариоцитах костного мозга, хранится в α-гранулах тромбоцитов. Активация тромбоцитов в месте повреждения сосуда приводит к высвобождению ФФВ, обеспечивающего их адгезию к эндотелию и агрегацию через гликопротеиды lb и llb/llla соответственно. Зрелый ФФВ содержит участки специфического связывания с разными лигандами и стабилизирует циркулирующий фактор VIII.
Обычно сопровождается умеренными кровотечениями. Скрининг-тесты показывают нормальное количество тромбоцитов и немного увеличенное АЧТВ. Диагноз ставится на основании низкого уровня антигена ФВ и нарушенной активности кофактора ристоцетина. Лечение включает в себя контроль за кровотечениями совместно с заместительной терапией или десмопрессином.
Уровень ФВ может временно возрастать при стрессе, физических упражнениях, беременности, воспалениях или инфекциях.
Выделяют 3 типа болезни Виллебранда.
- Тип 1: количественный дефицит ФВ, который является наиболее распространенной формой и аутосомно-доминантным заболеванием.
- Тип 2: качественные нарушения в синтезе ФВ, которые могут возникнуть в результате различных генетических аномалий. Является аутосомно-доминантным заболеванием.
- Тип 3: редкое аутосомно-рецессивное нарушение, при котором в гомозиготах не определяется ФВ.
БВ, как и гемофилия А, является наследственным заболеванием, которое в тяжелой форме может вызвать дефицит фактора VIII. Но этот дефицит, как правило, является умеренным.
Диагностика
 Заподозрить болезнь Виллебранда можно по характерной клинике и по семейному анамнезу. Для этого врачу необходимо провести тщательный расспрос пациента с детализацией жалоб, временем и частотой возникновения кровотечений и гематом, причинами возникновения симптомов. Обязателен и сбор анамнеза у близких родственников больного.
Заподозрить болезнь Виллебранда можно по характерной клинике и по семейному анамнезу. Для этого врачу необходимо провести тщательный расспрос пациента с детализацией жалоб, временем и частотой возникновения кровотечений и гематом, причинами возникновения симптомов. Обязателен и сбор анамнеза у близких родственников больного.
Из физикальных методов имеют значение проба жгута и щипка, которые позволяют наглядно продемонстрировать повышенную кровоточивость. На начальном этапе обследования помощь оказывает и общий анализ крови, имеющий данные о количестве эритроцитов и ретикулоцитов, об уровне гемоглобина и тромбоцитов. По этому стандартному анализу можно косвенно судить о тяжести и объеме кровопотери.
Более детальное изучение предполагаемой болезни потребует участия специального врача – гематолога, а также проведение более специфических тестов:
- развернутая коагулограмма;
- определение активности тромбоцитов;
- определение типа заболевания.
Внимание. Коагулограмма наиболее полно дает представление о состоянии свертывающей системы пациента
Время свертывания характеризует интервал от начала кровотечения до его полной остановки, при болезни Виллебранда оно значительно повышено.
Более четкие показатели дает определение протромбинового индекса, с помощью него можно спрогнозировать кровопотерю. Этапы коагуляции отражаются в анализе на АЧТВ и АВР, а уровень фибриногена характеризует возможности для образования нормального сгустка.
Активность тромбоцитарных телец определяют для выявления дефицита фактора VIII, для чего проводят реакцию с ристоцетином. Для диагностирования определенного типа болезни сравнивают агрегационную способность тромбоцитов пациента с нормальными показателями. Особую роль в определении формы патологии играет коллагеносвязывающая методика.
Книги
- Вилли Брандт Мирная политика в Европе = Friedenspolitik in Europa. — М.: Прогресс, 1969. — 170 с. — Рассылается по специальному списку
- Вилли Брандт Воля к миру: перспективы политики. — М.: Прогресс, 1972. — 325 с. — Рассылается по специальному списку
- Вилли Брандт Организованное безумие. Гонка вооружений и голод в мире = Der organisierte Wahnsinn. — М.: Прогресс, 1986. — 195 с. — Рассылается по специальному списку
- Вилли Брандт Права человека — поруганные и преступно нарушаемые = Menschenrechte Mißhandelt und Mißbraucht Reinbek. — М.: Прогресс, 1988. — 92 с. — Рассылается по специальному списку
- Вилли Брандт Воспоминания. — Новости, 1991. ISBN 5-7020-0153-2
- Вилли Брандт Демократический социализм. Статьи и речи. — Республика, 1992. ISBN 5-250-01286-8
- Вилли Брандт, Генри Киссинджер, Валери Жискар д’Эстен Брежнев. Уйти вовремя. — Алгоритм, 2012. ISBN 978-5-4438-0120-9
Биография
Герберт Эрнст Карл Фрам, будущий Вилли Брандт, родился в вольном городе Любеке (Германская империя); своего отца Йона Меллера Вилли Брандт не знал, воспитывался матерью — Мартой Фрам, работавшей продавщицей в кооперативном магазине, — и дедом, водителем грузовика и ветераном социалистического движения.
Свою первую политическую статью он написал ещё в тринадцать лет. В 1932 году окончил гимназию; ещё гимназистом в 1929 году вступил в организацию социалистической молодежи («Красные соколы»), годом позже стал членом Социал-демократической партии Германии (СДПГ). В 1931 году перешёл в более левую Социалистическую рабочую партию (СРП).
В 1933 году после прихода Гитлера к власти СРП была запрещена. Перейдя на нелегальное положение, партия продолжала бороться с нацистами, и Фраму, ставшему подпольщиком, было поручено создать филиал подпольной организации в Осло. В апреле 1933 года через Данию он эмигрировал в Норвегию, где изучал историю в Университете Осло. Тогда же взял себе псевдоним Вилли Брандт, под которым стал официально работать с 1947 года. В 1934 году принял участие в создании Международного бюро революционных молодёжных организаций и был избран в его секретариат от СРП.
В сентябре-декабре 1936 года Брандт по заданию руководителя подполья СРПГ в Париже Якоба Вальхера, выдавая себя за норвежского студента по имени Гуннар Гаасланд, курсировал между Норвегией и Германией для налаживания связей антифашистского подполья. В январе 1937 года от имени Социалистической рабочей партии вместе с лидерами КПГ и СДПГ, а также известными деятелями культуры подписал антифашистское воззвание к немецкому народу. В 1937 году в течение пяти месяцев в качестве военного корреспондента норвежских газет освещал события Гражданской войны в Испании.
В 1938 году Брандт был лишён гражданства немецким нацистским правительством, просил о предоставлении норвежского гражданства. Во время немецкой оккупации Норвегии в 1940 году попал в плен, но поскольку носил норвежскую военную форму, не был разоблачен и после кратковременного задержания в лагере для военнопленных смог бежать в Швецию в июне 1940 года. В августе 1940 года посольство в Стокгольме предоставило ему норвежское гражданство.
В Стокгольме Брандт оставался до конца войны, активно участвовал в организациях социал-демократов — политэмигрантов из европейских стран. Кроме того, он создал там «Шведско-норвежское бюро прессы», передававшее в международные СМИ информацию о положении дел в Германии и оккупированных странах, а также поддерживал связи со спецслужбами союзников. Брандт знал о покушении на Гитлера 20 июля 1944 года от немецкого дипломата, тайно участвовавшего в Сопротивлении, и одним из первых написал книгу об этом событии. В 1944 году вновь вступил в СДПГ.
После окончания Второй мировой войны Брандт вернулся в 1945 году в Германию в качестве корреспондента скандинавских газет, поселившись в западном секторе Берлина; в 1946 году занял должность пресс-атташе норвежской военной миссии в Германии. Лишь в 1948 году он вновь принял немецкое гражданство. В 1949 году полицейский президиум в Берлине утвердил его псевдоним Вилли Брандт в качестве официального имени.
7 декабря 1970 года в Варшаве канцлер Федеративной Республики Германия Вилли Брандт, находившийся с государственным визитом в Польше, преклонил колени перед памятником героям и жертвам Варшавского гетто. Столь необычным для государственного политика жестом он, боровшийся против нацизма и рисковавший жизнью в борьбе с ненавистным режимом, просил прощения от лица миллионов немцев за его преступления против еврейства.
За время же многочисленных визитов в СССР он ни в чём подобном замечен не был.
Симптомы
Основной симптом заболевания – обширные кровотечения различной формы. Это могут быть как легкие подкожные кровоизлияния и носовые кровотечения, так и продолжительные и обильные разной локализации. Также могут наблюдаться большие кровоизлияния и гематомы во внутренних органах и мягких тканях.
 Кроме того, эта патология может проявляться в виде маточного и носового кровотечения. Реже возникают внутричерепные или желудочно-кишечные. Очень редко наблюдается гемоартроз, при котором поражаются единичные суставы, что практически не влияет на работу опорно-двигательного аппарата.
Кроме того, эта патология может проявляться в виде маточного и носового кровотечения. Реже возникают внутричерепные или желудочно-кишечные. Очень редко наблюдается гемоартроз, при котором поражаются единичные суставы, что практически не влияет на работу опорно-двигательного аппарата.
Симптомы при легкой форме болезни: кровоточивость десен, регулярные кровотечения из носа, обильные и продолжительные менструации, беспричинное возникновение гематом.
При серьезных формах могут проявляться следующие симптомы:
- появление крови в моче (гематурия);
- синяки появляются даже при незначительных ушибах;
- стул темный смолистый с примесями крови;
- редко возникают внутрисуставные кровотечения, которые вызывают боль, затруднение движений и отечность.
Самые тяжелые и мучительные симптомы – миноррагии. Чрезмерно обильные и длительные менструации, которые приводят к анемии. Этот симптом является показанием для удаления матки.